Quick Link: Guideline | Gap Assessment | Implementation
EU MDR implementation guide for medical devices
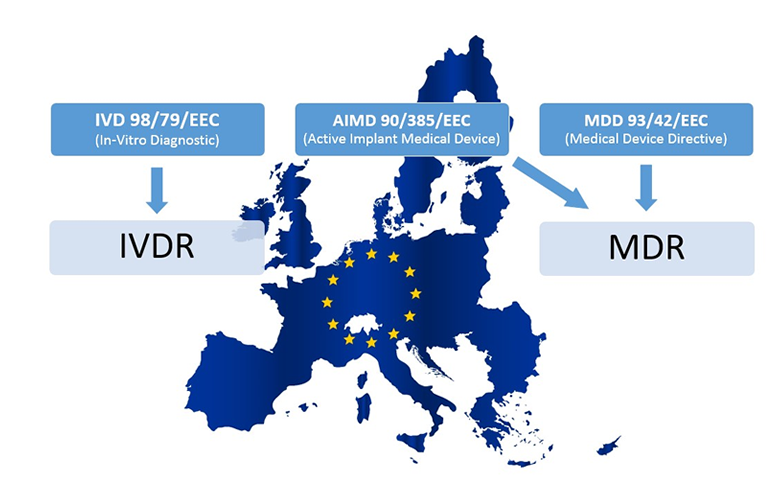
Medical Device Regulation (MDR)
The European medical device industry is undergoing a transformative phase due to the enactment of the new Medical Device Regulation (EU MDR), which was officially signed on May 26, 2017. Unlike its predecessor, the Medical Devices Directive (MDD), the EU MDR is a regulation, signifying a binding legislative act across all EU member states. This mandates all medical device companies marketing their products in Europe to comply with the new set of standards outlined in the EU MDR.
The enforcement deadline for compliance was May 26, 2021. Post this date, companies failing to adhere to the EU MDR will be prohibited from selling their medical products within the European Union. The extent of the impact brought about by this regulation is substantial and varies based on your designation as an economic operator, be it a manufacturer, importer, authorized representative, or distributor.
This shift aims to bolster the assurance of medical device safety and effectiveness while fostering innovation and competitiveness within the European medical device market. The following guide aims to provide a clear pathway towards achieving compliance with the EU MDR, ensuring a smooth transition for all stakeholders involved.
MDR Implementation Guide:
Embark on a structured journey towards compliance with the new Medical Device Regulation (MDR EU2017/745) through our step-by-step guide. This guide is designed to provide a simplified understanding, aiding in efficient and cost-effective implementation of the new regulation.
Disclaimer: This guide is based on our experiences and should not be taken as professional advice.
Preliminary Step: Engage with your Notified Body:
Confirm if your Notified Body is MDR certified and supports your device scope. Find the list of all MDR-certified Notified Bodies including information on their scope under the following link.
Yes: Schedule your MDR audit date!
No: Transition to a certified Notified Body promptly.
1.) Device Classification:
Review the new MDR rules to ascertain any impact on your current or future product classification. See MDR Annex XIII or use our classification form.
2.) Economic Operators:
Identify your role(s) as an economic operator(s) – Manufacturer, Importer, Distributor, or Authorized Representative, each having distinct MDR requirements.
Utilize our Economic Operators Tool for a clearer understanding of each role and its responsibilities.
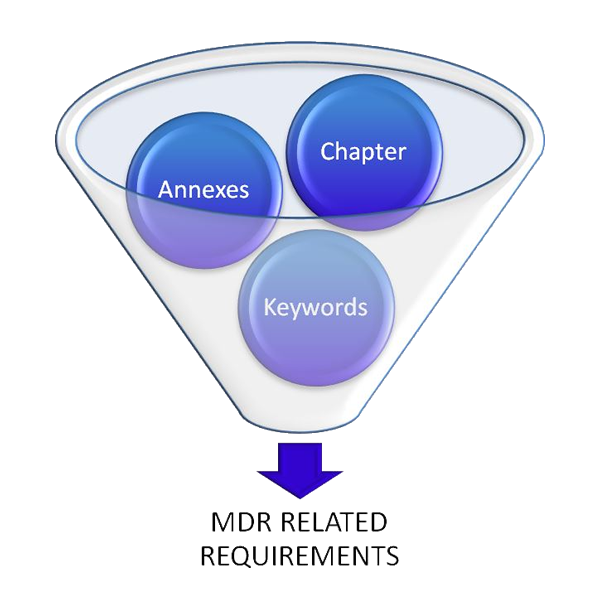
3.) Gap Assessment:
Utilize our MDR Gap-Analysis Tool to:
1. Step
Eliminate irrelevant information from the chapters and annexes.
2. Step
Identify and filter out non-applicable keywords.
3. Step
Review all requirements to determine their relevance to your business.
With our MDR Gap-Analysis Tool you are able to do all these steps above and you can reduce the information immediately.
4.) Implementation:
Prior to starting the implementation phase, you should put a plan in place. The steps below will guide you through the main topics. Do not forget to read MDR article 120 (transition period) carefully.
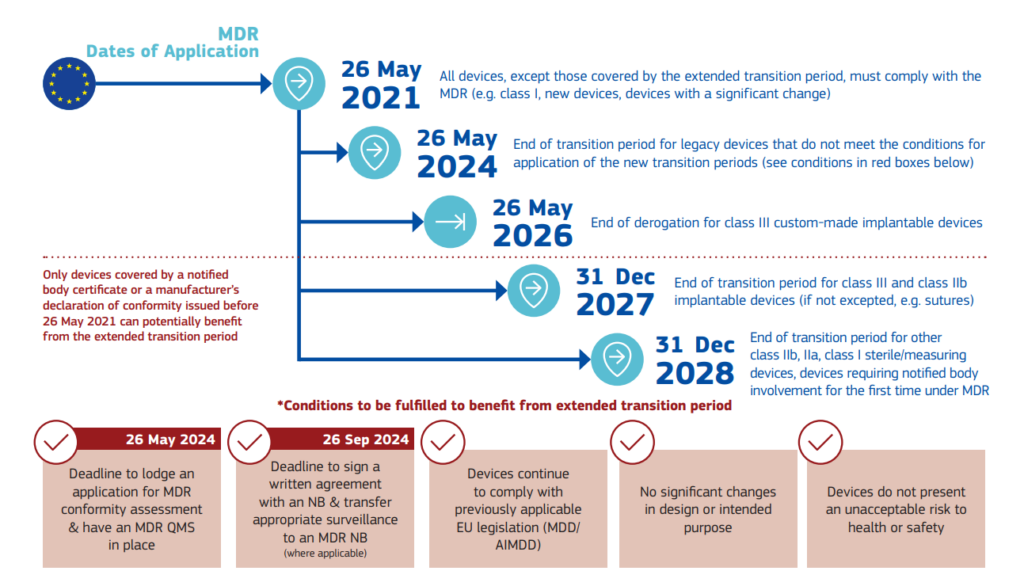
Update of the transition timelines according to the new Regulation (EU) 2023/607:
The main changes are:
EU MDR Transition Timelines (15th March 2023):
-until 31st December 2027 for Class IIb and III
-until 31ste December 2028 for Class I and IIa
-Until 26th May 2026 for Class III Implantable customer-made devices
-Extends validity of certificates issued up to 26 May 2021
-Remove “sell-off” date
These extended transition periods are intended to provide manufacturers with additional time to meet the new requirements of the EU MDR 2017/745. However, it’s important to note that these extensions apply only to certain medical devices, and manufacturers should carefully review the regulation to determine the specific deadlines applicable to their products.
During the transition period, devices that are compliant with the previously applicable Directives (AIMDD, MDD and IVDD) and devices that are compliant with the current Regulations (MDR and IVDR) can co-exist and may simultaneously be placed or made available on the EU market. This is particularly important for those third countries that rely on the CE marking of devices to grant access to their markets.
In conclusion, the transition period and deadlines for the EU MDR 2017/745 are critical aspects that manufacturers must understand and plan for. By doing so, they can ensure a smooth transition to the new regulatory framework, maintain market access, and ultimately ensure the safety and performance of their medical devices.
Source: Regulation (EU) 2023/607
4.1 Safety and performance checklist:
Our general safety and performance checklist stores the complete requirements of MDR annex 1.
4.2 Technical File:
Technical file update according to the MDR annex II requirements.
4.3 Risk management according to ISO 14971:
Your risk management activities (plan, analysis, and report) should be aligned with your PMS and PMCF activities.
4.4 UDI System:
Each medical device needs a UDI-DI (Unique Device Identification – Device Identifier) and UDI-PI (Unique Device Identification – Production Identifier) and must be submitted and transferred to the UDI database. (See document from the EU Commission)
MDCG documents:
MDCG 2018-1 Rev.4: Guidance on basic UDI-DI and changes to UDI-DI
MDCG 2018-3 Rev.1: Guidance on UDI for systems and procedure packs
MDCG 2018-4: Definitions/descriptions and formats of the UDI core elements for systems or procedure packs
MDCG 2018-5: UDI assignment to medical device software
MDCG 2018-6: Clarifications of UDI related responsibilities in relation to article 16
MDCG 2018-7: Provisional considerations regarding language issues associated with the UDI database
MDCG 2019-1: MDCG guiding principles for issuing entities rules on basic UDI-DI
MDCG 2019-2: Guidance on application of UDI rules to device-part of products referred to in article 1(8), 1(9) and 1(10) of Regulation 745/2017
MDCG 2020-18: MDCG Position Paper on UDI assignment for Spectacle lenses & Ready readers
MDCG 2021-9: MDCG Position Paper on the Implementation of UDI requirements for contact lenses, spectacle frames, spectacle lenses & ready readers
MDCG 2021-10: The status of Appendixes E-I of IMDRF N48 under the EU regulatory framework for medical devices
MDCG 2021-19: Guidance note integration of the UDI within an organisation’s quality management system
MDCG 2022-7: Q&A on the Unique Device Identification system under Regulation (EU) 2017/745 and Regulation (EU)
4.5 Post Market Surveillance (PMS):
Post market surveillance is defined in chapter VII of the MDR. Helpful MDCG document are:
MDCG 2022-21: Guidance on Periodic Safety Update Report (PSUR) according to Regulation (EU) 2017/745
MDCG 2023-3: Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices
MDCG 2024-1: Device Specific Vigilance Guidance (DSVG) Template
MDCG 2024-1-1: DSVG 01 on Cardiac ablation
MDCG 2024-1-2: DSVG 02 on Coronary stents
MDCG 2024-1-3: DSVG 03 on Cardiac implantable electronic devices (CIEDs)
MDCG 2024-1-4: DSVG 04 on Breast implants
MDCG 2024-1-5: DSVG 05 on Urogynaecological Surgical Mesh Implants used for Pelvic Organ Prolapse repair and Stress Urinary Incontinence
4.6 Post Market Clinical Follow-Up (PMCF):
Manufacturers shall conduct a clinical evaluation in accordance with the requirements set out in Article 61 and Annex XIV, including a PMCF. We have prepared a PMCF-Plan template.
4.7 Clinical Evaluation:
According to MEDDEV 2.7.1 Rev.4.
MDCG documents for clinical investigation and evaluation:
MDCG 2019-9 Rev.1: Summary of safety and clinical performance
MDCG 2020-5: Guidance on clinical evaluation – equivalence
MDCG 2020-6: Guidance on sufficient clinical evidence for legacy devices
MDCG 2020-7: Guidance on PMCF plan template
MDCG 2020-8: Guidance on PMCF evaluation report template
MDCG 2020-10/1 Rev.1: Guidance on safety reporting in clinical investigations
MDCG 2020-10/2 Rev.1: Guidance on safety reporting in clinical investigations
MDCG 2020-13: Clinical evaluation assessment report template
MDCG 2021-6 Rev.1: Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation
MDCG 2021-8: Clinical investigation application/notification documents
MDCG 2021-20: Instructions for generating CIV-ID for MDR Clinical Investigations
MDCG 2021-28: Substantial modification of clinical investigation under Medical Device Regulation
2023/C 163/06: Commission Guidance on the content and structure of the summary of the clinical investigation report
MDCG 2023-7: Guidance on exemptions from the requirement to perform clinical investigations pursuant to Article 61(4)-(6) MDR and on sufficient levels of access’ to data needed to justify claims of equivalence
MDCG 2024-3: Guidance on content of the Clinical Investigation Plan for clinical investigations of medical devices
MDCG 2024-5: Guidance on the Investigator’s Brochure content
MDCG 2024-10: Clinical evaluation of orphan medical devices
4.8 Labelling:
Manufacturers shall ensure that the device is accompanied by the information set out in Section 23 of Annex I in an official Union language(s) determined by the Member State in which the device is made available to the user or patient. The particulars on the label shall be indelible, easily legible, clearly comprehensible to the intended user or patient.
4.9 EUDAMED registration:
EUDAMED is now online but not all six modules are fully functional. It can be expected that the EUDAMED will be fully functional around 2029. More detailed information about EUDAMED and how it works can be found in our following blog post.
MDCG document for EUDAMED:
MDCG 2019-4: Timelines for registration of device data elements in EUDAMED
MDCG 2019-5: Registration of legacy devices in EUDAMED
MDCG 2020-15: Position Paper on the use of the EUDAMED actor registration module and of the single registration number (SRN) in the Member States
MDCG 2021-1 Rev.1: Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional
MDCG 2021-13 Rev.1: Questions and answers on obligations and related rules for the registration in EUDAMED of actors other than manufacturers, authorised representatives and importers subject to the obligations of Article 31 MDR and Article 28 IVDR
MDCG 2022-12: Guidance on harmonised administrative practices and alternative technical solutions until Eudamed is fully functional (for Regulation (EU) 2017/746 on in vitro diagnostic medical devices)
4.10 Common Specifications:
Take care about upcoming specification updates. We keep you informed with our Regulatory Intelligence Paper. Furthermore, this paper helps to address regulatory activities according to ISO 13485:2016, Chapter 5.6 Management Review. Detailed information can also be found here.
5.) Verification / Improvement
Performing internal audits and a final mock audit to ensure the key requirements have been implemented.
6.) Final Check List:
The Final checklist will ensure the completeness of your implementation process.
FAQ:
- What is EU MDR 2017/745?
The EU MDR 2017/745 is a regulation by the European Parliament and the Council that provides a regulatory framework for medical devices. It sets high standards of quality and safety for medical devices to meet common safety concerns. - What is the purpose of EU MDR 2017/745?
The purpose of the EU MDR 2017/745 is to harmonize the rules for the placing on the market and putting into service of medical devices and their accessories on the Union market, allowing them to benefit from the principle of free movement of goods. - What are the key changes in EU MDR 2017/745 compared to the previous directive (MDD)?
The EU MDR 2017/745 introduces several changes, including a new classification system for medical devices, increased post-market surveillance, a new Unique Device Identification (UDI) system, and stricter requirements for clinical evidence, among others. - What is the implementation date for EU MDR 2017/745?
The EU MDR 2017/745 came into effect on May 26, 2021. - What is the role of a Notified Body in the context of EU MDR 2017/745?
A Notified Body is an organization designated by an EU country to assess the conformity of certain products, including medical devices, before being placed on the market. These bodies carry out tasks related to conformity assessment procedures set out in the applicable legislation, including the EU MDR 2017/745. - What is the European Medical Device Nomenclature (EMDN)?
The EMDN is a system of internationally agreed terms used to identify and categorize medical devices. It is used in the context of EU MDR 2017/745 for the registration of medical devices in the EUDAMED database. - What is the impact of EU MDR 2017/745 on Class I medical devices?
Under the EU MDR 2017/745, Class I medical devices, which were previously self-certified, may now require a Notified Body review if they are sterile (Is), have a measuring function (Im), or are reusable surgical instruments (Ir). - What is the MDR’s stance on software as a medical device (SaMD)?
The MDR has specific rules for software. Depending on its intended use, software can be classified as a medical device (SaMD) and must comply with the relevant requirements of the MDR. - What are the post-market surveillance requirements under the EU MDR 2017/745?
The EU MDR 2017/745 requires manufacturers to actively and systematically gather, record and analyze relevant data on the quality, performance and safety of a device throughout its entire lifecycle, and to use the findings to take corrective or preventive actions. - What is the role of the ‘Person Responsible for Regulatory Compliance’ under the EU MDR 2017/745?
The Person Responsible for Regulatory Compliance (PRRC) ensures the conformity of the devices in terms of the MDR requirements. They are also responsible for the post-market surveillance, reporting obligations, and for keeping the technical documentation and the EU declaration of conformity up to date. - What are the labelling requirements under the EU MDR 2017/745?
The EU MDR 2017/745 has specific requirements for the labelling of medical devices. These include the need for a Unique Device Identification (UDI), the name and address of the manufacturer, information needed for safe use of the device, and any warnings or precautions to be taken. If the device is intended for clinical investigations, this must be clearly stated on the label. The label must also include any necessary information to identify the device, such as a device identifier or a production identifier. The information on the label must be presented in a way that is understood by the intended user and must be in the language(s) determined by the Member State in which the device is sold.
Glossary of Terms:
- Device Classification: The categorization of medical devices based on their risk profile, as defined by the MDR.
- Economic Operators: Entities involved in the production, distribution, or use of medical devices, including manufacturers, importers, distributors, and authorized representatives.
- EUDAMED: The European Database on Medical Devices, a secure web-based portal for data exchange between national competent authorities and the European Commission.
- Gap Assessment: An analysis to identify the differences between current operations and required compliance standards under the MDR.
- Implementation Plan: A structured approach detailing the steps and timelines to achieve compliance with the MDR.
- Medical Device Regulation (MDR EU2017/745): The regulatory framework governing the manufacture and distribution of medical devices within the European Union.
- Notified Body: An organization designated by an EU Member State to assess the conformity of certain products before being placed on the market.
- Post Market Clinical Follow-Up (PMCF): A continuous process to collect and evaluate clinical data from the use of a marketed device to ensure its continued safety and performance.
- Post Market Surveillance (PMS): The systematic collection, analysis, and interpretation of data related to the marketed device to identify any adverse events or risks associated.
- Vigilance: The process of identifying, evaluating, and controlling the risks related to the use of a medical device to protect the health and safety of patients, users, and others.
- Clinical Evaluation: A systematic process to collect, appraise, and analyze clinical data pertaining to a medical device to verify its safety and performance.
- Technical File: A document detailing the specifications, testing, and compliance information of a medical device.
- Unique Device Identification (UDI): A system of assigning a unique identifier to medical devices for tracking and monitoring purposes.
- UDI Database (UDID): A database that stores the UDI and other device information, facilitating the traceability of medical devices.
- European Medical Device Nomenclature (EMDN): A system of internationally agreed terms used to identify and categorize medical devices.
- ISO 13485: An internationally recognized standard that specifies requirements for a quality management system where an organization needs to demonstrate its ability to provide medical devices and related services that consistently meet customer and applicable regulatory requirements.
- Risk Management: The systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, controlling, and monitoring risk.
- Software as a Medical Device (SaMD): Software intended to be used for one or more medical purposes without being part of a hardware medical device.
- CE Marking: A certification mark that indicates conformity with health, safety, and environmental protection standards for products sold within the European Economic Area (EEA).
- Authorized Representative (AR): An entity designated by a manufacturer to act on their behalf in the European Union, responsible for ensuring compliance with regulatory requirements.
- Declaration of Conformity: A formal declaration by a manufacturer, or the manufacturer’s representative, that the product to which it applies meets all relevant provisions of all product safety directives applicable to that product.
- MDCG: Medical Device Coordination Group