30 Nov EUDAMED

EUDAMED and all you need to know
EUDAMED, an abbreviation of the European Database on Medical Devices, is an initiative developed by the European Commission. Its primary purpose is to enhance market transparency and oversight in the medical device field by providing a comprehensive repository of information on medical devices and their respective manufacturers active within the EU.
EUDAMED is more than just a repository, it’s a multi-faceted tool that integrates various processes related to the medical device lifecycle. It encompasses aspects such as product registration, post-market surveillance, vigilance, clinical investigations, and certification activities. Therefore, if you’re a manufacturer, understanding EUDAMED registration is not just essential—it’s a legal requirement.
How does EUDAMED registration work?
To start, it’s important to understand that EUDAMED is a cornerstone of the EU’s regulatory framework for medical devices, including the Medical Devices Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR). These regulations mandate the registration of all medical devices and their manufacturers in the EUDAMED database.
The EUDAMED registration process involves several steps. A manufacturer or their authorized representative must first apply for a Single Registration Number (SRN). This SRN is a unique identifier used across the EU to identify the manufacturer in the EUDAMED system. Once the SRN is assigned, manufacturers can then enter relevant device data into the system.
Manufacturers are required to provide detailed information about their devices, including the product’s unique device identification (UDI), details of clinical investigations, any conformity assessment procedures, certificates issued, incidents and corrective actions, and details of any recalls or withdrawals. Once entered into EUDAMED, this data is accessible to competent authorities, notified bodies, economic operators, sponsors, and the public, as appropriate.
Implications of EUDAMED for Different Types of Users
EUDAMED has significant implications for various stakeholders in the medical device industry. Here’s how it affects different types of users:
Manufacturers:
- Must register their company and obtain a Single Registration Number (SRN) (Only for companies based in EU)
- Required to input and maintain detailed information about their devices, including UDI data
- Responsible for reporting incidents, field safety corrective actions, and clinical investigations
- Need to ensure compliance with MDR/IVDR requirements through EUDAMED
- May need to invest in IT infrastructure and training to manage EUDAMED data effectively
Authorized Representatives:
- Must register in EUDAMED on behalf of non-EU manufacturers and obtain an SRN
- Responsible for verifying that manufacturers have uploaded required information
- Need to maintain up-to-date mandates and contact information in the system
- Act as a liaison between manufacturers and competent authorities
Importers:
- Required to register in EUDAMED and obtain an SRN
- Must verify that manufacturers have complied with EUDAMED registration requirements
- Responsible for reporting serious incidents and non-conformities to manufacturers and competent authorities
- Need to maintain records of complaints and device recalls
Notified Bodies:
- Use EUDAMED to issue and manage certificates
- Required to input information about conformity assessments and audits
- Must update certificate statuses and report any suspended or withdrawn certificates
- Responsible for monitoring manufacturers’ compliance with regulatory requirements
Competent Authorities:
- Use EUDAMED for market surveillance activities
- Access to comprehensive data for monitoring device safety and performance
- Responsible for validating actor registrations within their jurisdiction
- Coordinate with other authorities on vigilance and market surveillance activities
Healthcare Professionals and Patients:
- Gain access to more transparent information about medical devices
- Can search for device safety information and clinical data
- Ability to report incidents directly through EUDAMED
For all users, EUDAMED represents a significant shift towards greater transparency and data centralization in the medical device industry.
The six EUDAMED modules
These modules cover different aspects of the medical device lifecycle, facilitating both regulatory compliance and surveillance. The six modules are as follows:
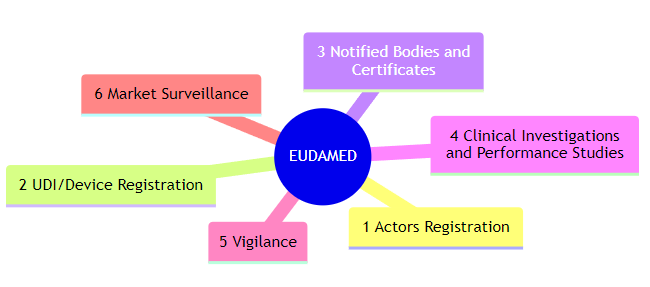
1. Actors Registration:
This module is used for the registration of manufacturers, authorized representatives, and importers. It provides each registered entity with a Single Registration Number (SRN). The following document describes in more detail how it works.
EUDAMED user guide Economic Operators – Actor module. Link to document
Here are some of the main topics:
- It provides instructions on how to log in and register as an actor
- The guide provides instructions on how to enter the required data, such as identifying your authorized representative, entering the validity start and end dates of the written mandate with the Authorised Representative, and uploading the summary mandate document.
- It also explains the user profiles per actor and their hierarchy.
- The guide provides information on how to handle different scenarios such as incomplete or incorrect information, wrong Competent Authority and/or Authorised Representative, duplicate actors, non-applicable requests, and suspected fraud. It also explains how to complete an assessment and search and view registered actors.
- It provides instructions on how to find your Authorised Representative from the search page, enter the Actor ID/SRN, specify the mandate’s validity start-date and end-date, and upload your summary mandate document.
- The guide explains how to view the details for any actor, navigate between the different information related to the actor, and search for an Authorised Representative.
Additional sources:
Notified Bodies Access User Guide – Link to document
Competent Authorities Actor Validation Guide – Link to document
Designated Authorities User Guide – Link to document
2. UDI/Device Registration:
The Unique Device Identification (UDI) and Device Registration module enables manufacturers to register their devices before placing them on the market. This includes details about the device and its classification. The following document describes in more detail how it works.
EUDAMED user guide UDI Devices – Production Link to document
Here are some of the main topics:
Basic UDI-DI Identification Details:
The guide provides instructions on how to fill in the Basic UDI-DI identification details. EUDAMED will validate the Basic UDI-DI code based on the specific format for each Issuing Entity and will prevent you from going further if the code is not valid. If the Basic UDI-DI code already exists in EUDAMED, the system will prevent you from saving, as a Basic UDI-DI must be unique.
Special Device Types:
The guide notes that it is currently not possible to register devices with certain special device types, including standard soft contact lenses, rigid gas permeable (RGP) contact lenses, made-to-order soft contact lenses, spectacle frames, spectacle lenses, and ready-made reading spectacles.
UDI-DI Code:
The guide provides instructions on how to enter the UDI-DI code, which must be a 14-digit code including the check digit that will be used by EUDAMED to validate the UDI-DI code. If the GS1 UDI-DI (GTIN code) is less than 14 digits, leading zeros should be added until it reaches 14 digits.
Managing Device Information:
The guide provides detailed instructions on how to manage device information, including how to delete a draft Basic UDI-DI/EUDAMED DI, how to update (create a new version) for Basic UDI-DI/EUDAMED DI, and how to view historical versions for Basic UDI-DI/EUDAMED DI.
Discarding Registered UDI-DIs/EUDAMED IDs:
The guide explains that the discard operation acts as a final deactivation. A device in state discarded is therefore not listed and cannot be viewed in the public site of EUDAMED. However, it can be viewed by the Manufacturer (owner of the discarded device), Competent Authority, and Notified Body actors.
3. Notified Bodies and Certificates:
This module contains information about Notified Bodies (organizations designated by EU member states to assess the conformity of certain products before being placed on the market) and the certificates they issue. The following document describes in more detail how it works.
EUDAMED user guide Notified Bodies & Certificates. Link to document
Here are some of the main topics:
- The guide provides instructions on how to review different versions of a document. You can click on the version you wish to review, which opens a summary of it.
- It also guides you on how to register a refused certificate. You can preview your choice or click submit.
- The guide explains how to register a withdrawn application. It mentions that you can click ‘Preview’ to double-check the content in each step.
- When you are done with certain tasks, you can click ‘Submit’. If a translation in English is not provided, the system will display a warning message that an English translation is required. It also explains how to view the SS(C)P version history.
- The guide provides instructions on how to download certificates and refused certificates in a structured format.
4. Clinical Investigations and Performance Studies:
The Clinical Investigations and Performance Studies module in EUDAMED is a crucial component for enhancing transparency and oversight of clinical research in the medical device (MD) and in-vitro diagnostic (IVD) industry. This module serves several important functions:
Purpose:
- To centralize information on clinical investigations for medical devices and performance studies for in vitro diagnostic devices (IVDs).
- To facilitate the coordination and information sharing between EU member states regarding these studies.
- To increase transparency of clinical data for healthcare professionals, patients, and the public.
Key Features:
- Study Registration: Manufacturers and sponsors must register their clinical investigations and performance studies before they begin.
- Application Management: The module supports the submission and management of applications for clinical investigations and performance studies.
- Multi-State Coordination: For studies conducted in multiple EU countries, the module facilitates coordinated assessment and decision-making.
- Progress Tracking: Users can update and track the progress of ongoing studies, including any modifications or early terminations.
- Results Reporting: Upon completion, study results must be uploaded to the system, promoting transparency and data sharing.
User Implications:
- Manufacturers and Sponsors: Must use this module to register studies, submit applications, and report results. This requires careful planning and timely data entry.
- Competent Authorities: Can use the module to review applications, coordinate with other member states, and monitor ongoing studies in their jurisdiction.
- Ethics Committees: May have access to relevant study information to support their review processes.
- Healthcare Professionals and Public: Can access non-confidential information about ongoing and completed studies, enhancing transparency in medical device research.
Compliance Requirements:
- Registration of clinical investigations is mandatory under the Medical Device Regulation (MDR) and for performance studies under the In Vitro Diagnostic Regulation (IVDR).
- Timelines for registration and reporting are specified in the regulations and must be strictly adhered to.
- Failure to comply with registration and reporting requirements can result in regulatory actions and impact the ability to market devices in the EU.
Integration with Other Modules:
- The Clinical Investigations and Performance Studies module is closely linked with the UDI/Device Registration module, as studies must be associated with specific devices or device groups.
- It also interfaces with the Vigilance module for reporting any serious adverse events that occur during clinical investigations.
Future Developments:
- As EUDAMED continues to evolve, this module may see enhancements in functionality, such as improved data analysis tools or integration with other international clinical trial databases.
By providing this comprehensive information in the Clinical Investigations and Performance Studies module, EUDAMED aims to streamline the clinical research process, enhance patient safety, and promote evidence-based decision-making in the medical device industry.
5. Vigilance:
The Vigilance module in EUDAMED is a critical component for enhancing patient safety and post-market surveillance of medical devices in the European Union. This module facilitates the reporting, evaluation, and dissemination of information about serious incidents and field safety corrective actions (FSCAs) associated with medical devices.
Key Features and Functions:
- Incident Reporting: Manufacturers can use this module to report serious incidents related to their devices within the specified timelines.
- FSCA Reporting: The module allows for the reporting of Field Safety Corrective Actions, including recalls and safety notifications.
- Trend Reporting: Manufacturers can report statistically significant increases in the frequency or severity of non-serious incidents or expected undesirable side-effects.
- Competent Authority Access: National competent authorities can access and analyze incident reports, facilitating coordinated responses across the EU.
- Information Sharing: The module enables sharing of safety-related information among manufacturers, competent authorities, and healthcare professionals.
- Public Access: Non-confidential information about incidents and FSCAs is made available to the public, enhancing transparency.
User Implications:
- Manufacturers: Must familiarize themselves with the reporting requirements and timelines. They need to ensure their internal processes align with EUDAMED’s Vigilance module for timely and accurate reporting.
- Authorized Representatives: May be responsible for reporting incidents on behalf of non-EU manufacturers.
- Competent Authorities: Use the module to monitor incidents, coordinate responses, and communicate with other EU member states.
- Healthcare Professionals: Can access safety information and may be required to report incidents through their national systems, which may interface with EUDAMED.
Compliance Requirements:
- Reporting timelines vary based on the severity of the incident, ranging from immediate reporting for serious public health threats to up to 15 days for other serious incidents.
- Manufacturers must provide initial and follow-up reports, as well as final reports once investigations are complete.
- Periodic summary reports may be required for similar incidents with known root causes.
Integration with Other Modules:
- The Vigilance module is closely linked with the UDI/Device Registration module, as incident reports must be associated with specific registered devices.
- It also interfaces with the Market Surveillance module, supporting coordinated actions by competent authorities.
Benefits:
- Enhances patient safety through rapid identification and communication of device-related risks.
- Improves transparency in the medical device market.
- Facilitates coordinated responses to safety issues across the EU.
- Provides valuable data for continuous improvement of device safety and performance.
By leveraging the Vigilance module in EUDAMED, stakeholders in the medical device industry can contribute to a more robust and responsive post-market surveillance system, ultimately enhancing patient safety and public health protection across the European Union.
6. Market Surveillance:
The Market Surveillance module in EUDAMED is a crucial component designed to enhance the coordination and effectiveness of market surveillance activities for medical devices across the European Union. This module facilitates information sharing and collaboration among competent authorities to ensure the safety and compliance of medical devices on the market.
Key Features and Functions:
- Information Sharing: Enables competent authorities to share data on non-compliant devices, including details of inspections, testing results, and enforcement actions.
- Coordination of Activities: Facilitates the coordination of market surveillance activities between different EU member states, helping to avoid duplication of efforts.
- Risk Assessment: Supports the assessment and communication of potential risks associated with specific devices or categories of devices.
- Reporting Capabilities: Allows for the generation of reports on market surveillance activities, trends, and outcomes.
- Alert System: Provides a platform for rapid communication of urgent safety concerns or non-compliance issues among authorities.
- Link to Other Modules: Integrates with other EUDAMED modules, particularly the UDI/Device Registration and Vigilance modules, for comprehensive oversight.
User Implications:
- Competent Authorities: Primary users of this module. They can input data on surveillance activities, access information from other member states, and coordinate joint actions.
- Manufacturers: While not direct users, manufacturers may be impacted by increased scrutiny and coordinated market surveillance activities.
- Economic Operators: Importers and distributors may be subject to more consistent and coordinated inspections across the EU.
- Healthcare Providers and Patients: Benefit from enhanced safety measures and more rapid responses to identified risks.
Key Processes:
- Planning and Coordination: Authorities can use the module to plan and coordinate surveillance activities, including joint inspections or testing programs.
- Data Entry and Reporting: Results of inspections, laboratory tests, and other surveillance activities are entered into the system.
- Non-Compliance Management: The module supports the tracking and management of non-compliance cases, including corrective actions and follow-ups.
- Trend Analysis: Authorities can analyze trends in non-compliance or emerging risks across device categories or manufacturers.
- Resource Allocation: Helps authorities to allocate resources more effectively based on risk assessments and historical data.
Benefits:
- Enhances the efficiency and effectiveness of market surveillance activities across the EU.
- Promotes a more uniform approach to enforcement of medical device regulations.
- Facilitates rapid response to safety issues through improved information sharing.
- Supports evidence-based decision-making in regulatory actions.
Challenges and Considerations:
- Requires consistent data entry and active participation from all member states to be fully effective.
- Necessitates training and adaptation for competent authorities to fully utilize the system’s capabilities.
- May lead to increased regulatory scrutiny for manufacturers and economic operators.
By leveraging the Market Surveillance module in EUDAMED, competent authorities can work more collaboratively and efficiently to ensure the safety and compliance of medical devices in the European market. This enhanced coordination contributes to a more robust and responsive regulatory environment, ultimately benefiting patient safety and public health across the EU.
EUDAMED Roadmap – By when EUDAMED is fully functional
The EU Commission has published a new Draft Roadmap with updated timelines.
These are the following changes:
- Mandatory use of the modules Actor, Certificates, MSU, Vigilances, CI/PS by Q4 2027 as per EU MDR article 123(3)(d) and EU IVDR article 113(3)(f).
- Use of EUDAMED for Devices and Certificates registration becomes mandatory by Q2 2029 as per EU MDR article 123(3)(e) and EU IVDR article 113(3)(a).
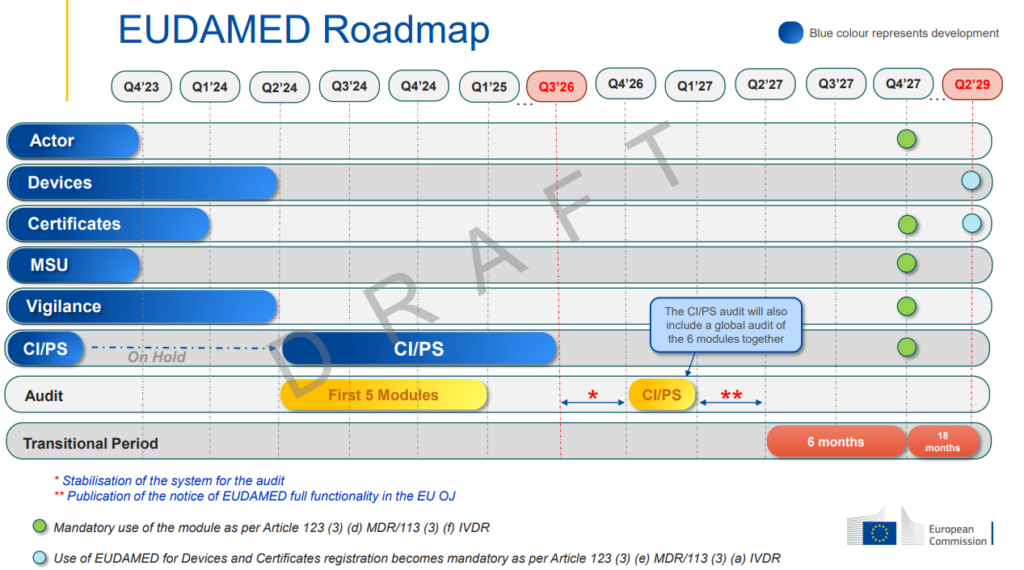
Updated in July 2024 – Gradual roll out and modules functionality view
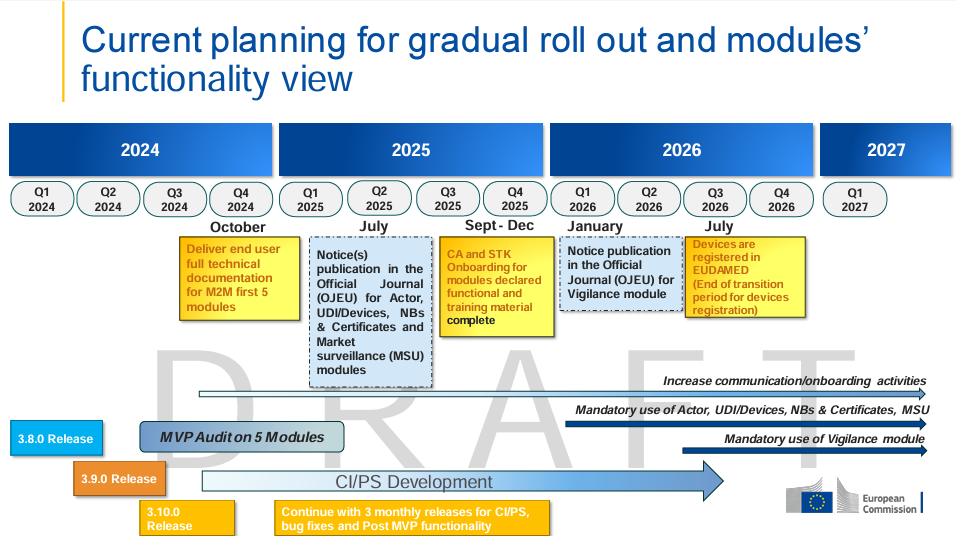
Conclusion: It is still a long way to go and it can be expected that EUDAMED will be fully functional somewhere around 2029.
Data Exchange with EUDAMED
EUDAMED offers three main methods to exchange data with EUDAMED. This are the methods:
- The User Interface (UI): This method involves manual data input through the EUDAMED application. It is the simplest form of data handling.
- The XML Bulk Upload/Download: This semi-automated option allows data to be uploaded in XML format. Users need to generate XML files, which must be validated against the provided EUDAMED DTX service and entity model XSDs. While the generation of these files can be automated, the uploading and downloading actions remain manual. This method is useful for bulk uploading of existing information and requires some implementation effort by an IT team.
- The Machine-to-Machine Data Exchange (M2M-DTX): This method enables automatic data exchange between an external backend system and EUDAMED’s backend services. It requires the external system to convert data into the XML format requested by EUDAMED DTX and to implement a specific data exchange protocol. This is the most complex and costly solution, suitable when there is a significant amount of data to be exchanged frequently, and when the cost of manual input outweighs the cost of automation.
The following documents describe in more detail how economic operators can exchange their data on EUDAMED.
Guidelines on Data Exchange with EUDAMED – Link to document
Machine-to-Machine Data Exchange – Link to document
Other helpful documents and sources:
EUDAMED public database – Link
EUDAMED information center – Link
MDCG 2022-12 – Guidance on harmonized administrative practices and alternative technical solutions until Eudamed is fully functional (for Regulation (EU) 2017/746 on in vitro diagnostic medical devices)
MDCG 2021-13 Rev. 1 – Questions and answers on obligations and related rules for the registration in EUDAMED of actors other than manufacturers, authorized representatives and importers subject to the obligations of Article 31 MDR and Article 28 IVDR
MDCG 2021-1 Rev. 1 – Guidance on harmonized administrative practices and alternative technical solutions until EUDAMED is fully functional
MDCG 2020-15 – MDCG Position Paper on the use of the EUDAMED actor registration module and of the Single Registration Number (SRN) in the Member States
MDCG 2019-5 – Registration of legacy devices in EUDAMED
MDCG 2019-4 – Timelines for registration of device data elements in EUDAMED
Regulatory Intelligence Service:
We are helping companies with being up to date with medical device regulation globally. Learn more and join our exclusive service by clicking HERE.
No Comments