Quick Link: Guideline | Gap Assessment | Implementation
IVDR implementation Guide
In-Vitro Diagnostic Regulation (IVDR)
The European medical device industry will undergo significant changes as a result of the new in-vitro diagnostic regulation which has signed on May 26th, 2017. As the name suggests, is it a regulation and no longer a directive and all in-vitro diagnostic companies that sell medical products in Europe have to adhere to this new regulation.
Companies that do not follow this regulation will no longer be allowed to sell their medical products in the European Union after May 26th, 2022. Depending on your role as economic operator (manufacturer, importer, authorized representative, distributor) the impact can be meaningful.
We, at Regulatory Globe, have developed a set of tools to help you solve all regulatory issues.
IVDR Implementation Guide:
Below you will find a step-by-step implementation guide with regards to the new in-vitro diagnostic regulation (MDR EU2017/746). Our guide is simple to understand and will allow you to save time and money when implementing the new regulation.
(Note: This guide should not be considered as a recommendation, it’s just based on our own experiences.)
Before you start, you should clarify the following with your Notified Body:
Will your Notified Body be certified by IVDR and your product categories are covered in their scope?
Yes: Save your IVDR audit date!
No: Change your Notified Body as soon as possible!!!
1.) Device Classification:
Check if the new IVDR rules have any impact on the classification of the existing or future products. The classification rules are defined in Annex VIII.
2.) Economic Operators:
Depending on your business you have one or multiple economic operator responsibilities, which is imperative to know before you start with the gap assessment. Economic operators are Manufacturers, Importers, Distributors or Authorized Representative. Each of them has different requirements to fulfill the IVDR.
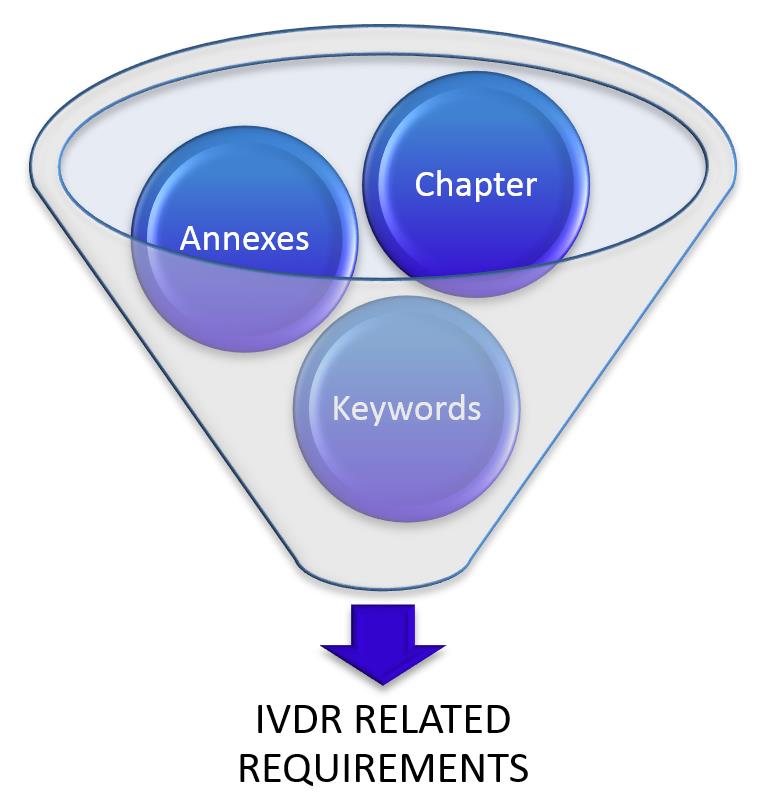
3.) Gap Assessment:
1. Step
Reduce not required information by going through the chapters and annexes and eliminate all not required information.
2. Step
Define keywords which are not applicable to you. In our IVDR tool you have the opportunity to search for these keywords.
3. Step
Go through all open requirements step by step and define if requirements are relevant for your business or not.
With our IVDR Gap-Analysis Tool you are able to do all these steps above and you can reduce the information immediately.
4.) Implementation:
Prior to starting the implementation phase, you should put a plan in place. The steps below will guide you through the main topics. Do not forget to read IVDR article 110 (transition provisions) in combination with EU 2024/1860 carefully.
![]()
UPDATE OF THE TRANSITION TIMELINES FOR DEVICES MEET CERTAIN CRITERIA ACCORDING TO THE NEW REGULATION (EU) 2024/1860:
The main changes are:
EU IVDR Transition Timelines (9th July 2024):
-until 31st December 2027 for Class D
-until 31st December 2028 for Class C
-Until 31st December 2029 for Class B and A (Steril condition)
The specific steps for a manufacturer to get an extended transitional period for their IVD products include ensuring that the devices are safe and taking certain steps to transition towards compliance with Regulation (EU) 2017/746. Manufacturers need to work with notified bodies to carry out the required conformity assessments within the extended transitional period. The extension aims to ensure a high level of public health protection, including patient safety, and to avoid shortages of IVD medical devices needed for healthcare services.
Furthermore, this EU Regulation 2024/1860, adopted in response to urgent public health concerns and delays in implementing the Eudamed electronic system for medical devices, aims to ensure the availability and safety of in vitro diagnostic (IVD) medical devices in the European market. It introduces measures to address potential shortages of IVD products, update certification processes, and provide clarity for manufacturers, economic operators, and healthcare providers. The Regulation includes provisions for gradual implementation of Eudamed, obligations for notifying interruptions or discontinuations of device supply, and transitional arrangements for certain IVD medical devices. Overall, the Regulation focuses on safeguarding patient health and enhancing the efficiency of the IVD market within the EU.
Source: Regulation EU 2024/1860
4.1 Safety and performance checklist:
Create a General Safety and Performance document according to the IVDR requirements in annex I.
4.2 Technical File:
Technical file update according to Annex II and III.
4.3 Risk management according to ISO 14971:
Your risk management activities (plan, analysis, and report) should be aligned with your PMS and PMPF activities.
4.4 UDI System:
Each medical device needs a UDI-DI (Unique Device Identification – Device Identifier) and UDI-PI (Unique Device Identification – Production Identifier) and must be submitted and transferred to the UDI database. (See document from the EU Commission).
4.5 Post Market Surveillance (PMS):
Post market surveillance is defined in chapter VII of the IVDR.
4.6 Post Market Performance Follow-Up (PMPF):
Manufacturers shall conduct a performance evaluation in accordance with the requirements set out in Article 56 and Annex XIII, including a PMPF.
4.7 Performance Evaluation Report (PER):
Create a performance evaluation report (PER) according to Annex XIII.
4.8 Labelling:
Manufacturers shall ensure that the device is accompanied by the information set out in Section 20 of Annex I in an official Union language(s) determined by the Member State in which the device is made available to the user or patient. The particulars on the label shall be indelible, easily legible and clearly comprehensible to the intended user or patient.
4.9 EUDAMED registration:
EUDAMED is now online but not all six modules are fully functional. It can be expected that the EUDAMED will be fully functional around 2029. More detailed information about EUDAMED and how it works can be found in our following blog post.
4.10 Common Specifications:
Take care about upcoming specification updates. We keep you informed with our Regulatory Intelligence Paper. Furthermore, this paper helps to address regulatory activities according to ISO 13485:2016, Chapter 5.6 Management Review.
5.) Verification / Improvement
Performing internal audits and a final mock audit to ensure the key requirements have been implemented.
6.) Final Check List:
The Final checklist will ensure the completeness of your implementation process.